Whether you’re at the reaction bench or reviewing a chapter on organic transformations, knowing common pathways makes planning and troubleshooting much easier. Seeing mechanisms side-by-side helps you predict products, choose reagents, and explain unexpected outcomes without guesswork.
There are 20 Types of Reaction Mechanisms, ranging from Allylic substitution (π-allyl complexes) to Sigmatropic rearrangements. For each entry you’ll find below data organized with Category,Key steps,Typical reagents/conditions so you can quickly compare scope and setup — you’ll find the full list and details below.
How can I tell which mechanism is operating in a given reaction?
Compare substrate structure, stereochemical outcomes, kinetics, and by-products; use simple experiments like rate dependence, isotope labeling, or trapping intermediates, and check whether observations match known patterns (for example, allylic systems often show π-allyl behavior while concerted shifts point to sigmatropic mechanisms).
What practical changes reliably steer a reaction toward a desired mechanism?
Adjust solvent polarity, temperature, catalysts/ligands, and concentration; try alternative leaving groups or additives, run small-scale trials with monitoring (TLC, NMR), and consult the table below for typical reagents and conditions that favor one pathway over another.
Types of Reaction Mechanisms
| Name | Category | Key steps | Typical reagents/conditions |
|---|---|---|---|
| SN2 | Ionic | Backside nucleophilic attack; concerted bond-making and bond-breaking, inversion of configuration | Strong nucleophiles, polar aprotic solvents, primary/secondary alkyl halides |
| SN1 | Ionic | Carbocation formation, nucleophilic attack, possible rearrangement or capture | Weak nucleophiles, polar protic solvents, tertiary alkyl halides, stabilizing groups |
| E2 | Ionic | Base abstracts β-hydrogen while leaving group departs in one concerted step | Strong bases, polar aprotic or protic solvents, antiperiplanar geometry required |
| E1 | Ionic | Formation of carbocation, β-hydrogen elimination to give alkene | Weak bases, polar protic solvents, tertiary substrates, heat often promotes |
| Nucleophilic acyl substitution | Ionic (acyl) | Nucleophilic addition to carbonyl, tetrahedral intermediate, leaving group departs | Nucleophiles (amines, alcohols), acid/base catalysis, acyl chlorides/esters, carboxylic derivatives |
| Electrophilic aromatic substitution (EAS) | Ionic (aromatic) | Aromatic electrophile forms σ-complex, deprotonation restores aromaticity | Strong electrophiles (Br+, NO2+), Lewis acids (FeBr3), activated aromatics |
| Nucleophilic aromatic substitution (SNAr) | Ionic (aromatic) | Addition to aromatic ring, Meisenheimer complex, leaving group departure | Strong nucleophiles, electron-withdrawing ring substituents, elevated temperatures |
| Benzyne (elimination-addition) | Ionic (aromatic) | Elimination forms benzyne intermediate; nucleophile adds to strained alkyne, protonation | Strong bases, high temperatures, ortho-leaving groups on aryl halides |
| Radical chain mechanism | Radical | Initiation, propagation, termination steps involving radicals | Radical initiators (peroxides), heat/UV, abstraction agents, radical traps |
| Photoredox (photoinduced electron transfer) | Radical/Photochemical | Photoexcited catalyst transfers electron, radical formation, subsequent bond-forming steps | Photocatalysts (Ru, Ir, organic dyes), visible light, sacrificial electron donors/acceptors |
| Diels-Alder (concerted cycloaddition) | Pericyclic | Concerted [4+2] cycloaddition between diene and dienophile | Electron-rich diene, electron-poor dienophile, heat or Lewis acid catalysis |
| Electrocyclic reactions | Pericyclic | Conrotatory or disrotatory ring opening/closure via concerted bond reorganization | Heat or light determines stereochemistry; conjugated polyenes substrates |
| Sigmatropic rearrangements | Pericyclic | Concerted shift of σ-bond with concomitant π-system reorganization | Thermal or photochemical activation; allylic or vinyl systems, heat |
| Cross-coupling catalytic cycle (Pd-catalyzed) | Catalytic (organometallic) | Oxidative addition, transmetalation, reductive elimination catalytic cycle | Palladium catalysts, organoboron/organostannane partners, bases, ligands |
| Allylic substitution (π-allyl complexes) | Catalytic (organometallic) | Formation of π-allyl intermediate, nucleophilic attack or substitution | Pd or other metal catalysts, allylic acetates/halides, nucleophiles |
| Michael (conjugate) addition | Ionic (conjugate addition) | Nucleophile adds to β-carbon of α,β-unsaturated carbonyl, protonation | Soft nucleophiles, bases or catalysts, enones/enals, organocatalysts or metals |
| Nucleophilic addition to carbonyls | Ionic | Nucleophile attacks carbonyl, tetrahedral alkoxide intermediate, protonation | Hydride donors, organometallics, cyanide, acids or bases for activation |
| General acid–base catalysis | Catalytic/Enzymatic | Proton transfer accelerates bond-making or bond-breaking steps | Acids/bases, enzymatic active-site residues, buffers, general acid or base catalysts |
| Enzymatic covalent catalysis | Enzymatic | Nucleophilic enzyme residue forms covalent intermediate, then hydrolysis regenerates enzyme | Active-site nucleophiles (Ser, Cys), cofactors, physiological pH |
| Heterogeneous catalytic hydrogenation | Catalytic (heterogeneous) | Adsorption of H2 and substrate on metal surface, hydrogen transfer, desorption | Supported metals (Pd, Pt, Ni), H2 gas, solvent, elevated pressures |
Images and Descriptions
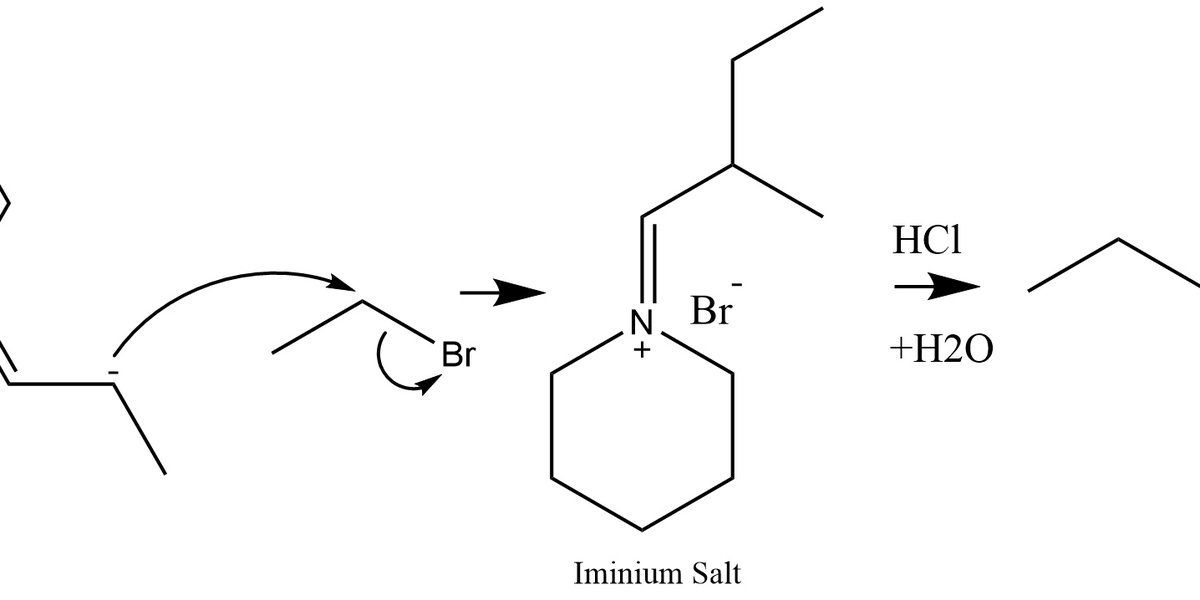
SN2
SN2 is a one-step substitution where a nucleophile attacks a saturated carbon as the leaving group departs simultaneously. It gives inversion of stereochemistry, favors strong nucleophiles and polar aprotic solvents, and is fastest at primary centers.
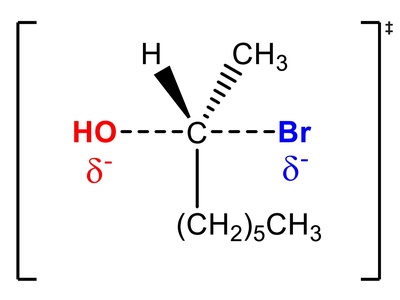
SN1
SN1 involves ionization to a carbocation intermediate, followed by nucleophile capture. Carbocation stability, rearrangements, and racemization are hallmarks. Favored by tertiary centers, polar protic solvents, and weak nucleophiles; kinetics show first-order dependence on substrate concentration.

E2
E2 is a concerted β-elimination where a base removes a hydrogen as the leaving group leaves, forming a double bond. Stereochemistry (antiperiplanar requirement) controls product geometry; favored by strong bases and secondary or tertiary substrates.
E1
E1 proceeds via carbocation formation, then deprotonation to form an alkene. Competes with SN1, shows Zaitsev’s rule preferences and possible rearrangements. Favored by stable carbocations, weak bases, and conditions that promote ionization.
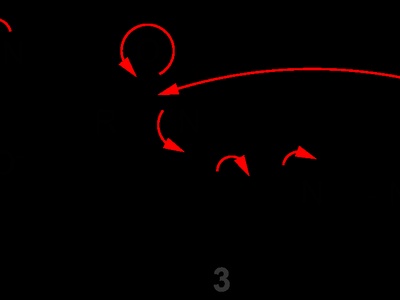
Nucleophilic acyl substitution
Nucleophilic acyl substitution involves nucleophile attack on a carbonyl forming a tetrahedral intermediate, then expulsion of a leaving group to replace the acyl substituent. It’s central to ester, amide, and acyl chloride chemistry and sensitive to leaving-group ability.
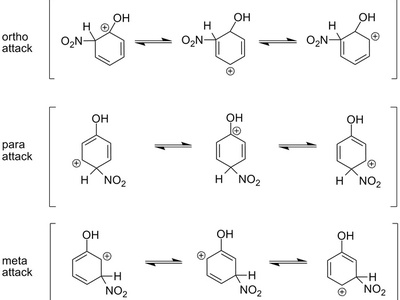
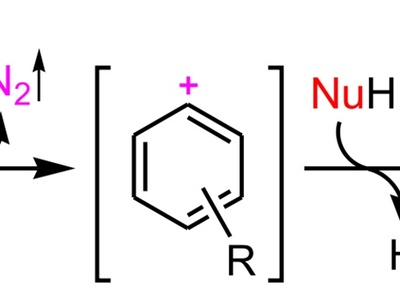
Electrophilic aromatic substitution (EAS)
EAS replaces an aromatic hydrogen by an electrophile via formation of a nonaromatic σ-complex (arenium ion) followed by deprotonation to regain aromaticity. Directing effects and stability of intermediates control regioselectivity; common in halogenation, nitration, and sulfonation.
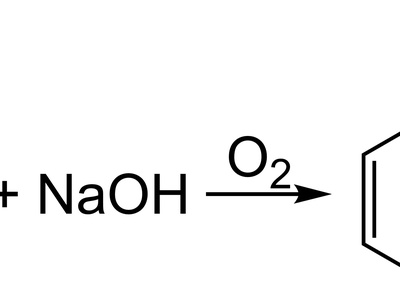
Nucleophilic aromatic substitution (SNAr)
SNAr proceeds by nucleophile addition to an electron-poor aromatic ring forming a Meisenheimer intermediate, then expulsion of a leaving group. It’s common on nitro-activated halobenzenes and contrasts with electrophilic aromatic substitution mechanisms.
Benzyne (elimination-addition)
The benzyne mechanism involves elimination to generate a strained triple-bond intermediate (benzyne), which is then attacked by nucleophiles. It leads to mixtures of regioisomers and typically requires harsh conditions or strong bases to form the reactive benzyne.

Radical chain mechanism
Radical chain reactions proceed via initiation (radical generation), propagation (chain-carrying steps that produce new radicals), and termination. Common in halogenation and polymerization, they show chain length effects and are sensitive to inhibitors and radical-stabilizing substituents.
Photoredox (photoinduced electron transfer)
Photoredox mechanisms use light-activated catalysts to perform single-electron transfers that create radical intermediates. These radicals enable unusual bond formations under mild conditions, enabling cross-couplings, oxidations, and reductions with high functional-group tolerance and often unique selectivity.
Diels-Alder (concerted cycloaddition)
The Diels–Alder reaction is a concerted pericyclic [4+2] cycloaddition forming six-membered rings with stereospecificity. It proceeds via a cyclic transition state, is thermally allowed, and is widely used for building complex ring systems in synthesis.
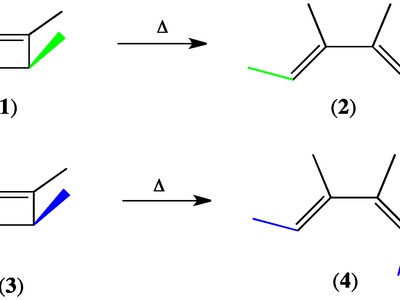
Electrocyclic reactions
Electrocyclic reactions convert conjugated polyenes between open-chain and cyclic forms through a concerted rotation of terminal bonds. Thermal versus photochemical conditions dictate conrotatory or disrotatory motion, giving predictable stereochemical outcomes based on orbital symmetry rules.
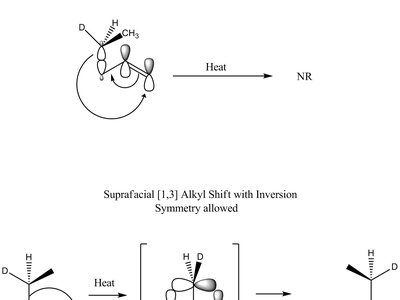
Sigmatropic rearrangements
Sigmatropic rearrangements move a σ-bond across a π-system in a single concerted step, changing connectivity without intermediates. Examples like Claisen and Cope rearrangements redistribute atoms predictably and are driven by orbital symmetry and the stability of transition states.
Cross-coupling catalytic cycle (Pd-catalyzed)
Cross-coupling catalytic cycles, typified by palladium catalysis, involve oxidative addition of an electrophile to metal, transmetalation with an organometallic partner, and reductive elimination to form a new carbon–carbon bond. Ligands, bases, and solvent control rates and selectivity.
Allylic substitution (π-allyl complexes)
Allylic substitution proceeds via formation of a π-allyl metal complex that stabilizes positive charge and allows nucleophilic attack at different positions. It explains regio- and stereoselectivity in Tsuji–Trost–type reactions and asymmetric catalysis.
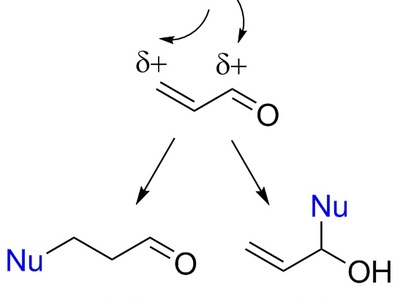
Michael (conjugate) addition
Michael addition is nucleophilic conjugate addition to α,β-unsaturated carbonyl compounds, forming new C–C or C–X bonds at the β-position. It often proceeds under base or catalysis and is a key C–C bond-forming strategy in synthesis.
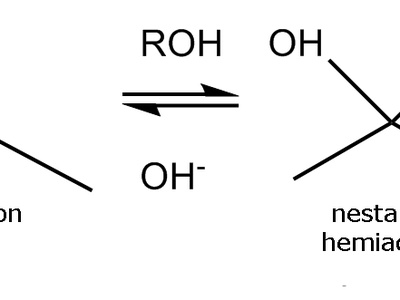
Nucleophilic addition to carbonyls
Nucleophilic addition to carbonyls converts carbonyls into alcohols or related derivatives via tetrahedral intermediates. Examples include addition of hydride, organometallic reagents, or cyanide; reactivity depends on carbonyl electrophilicity and the nucleophile’s strength.
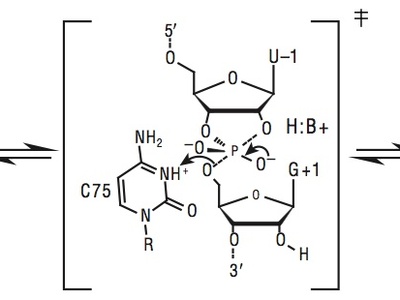
General acid–base catalysis
General acid–base catalysis uses proton donors or acceptors to lower activation barriers by stabilizing charged transition states. Common in both small-molecule catalysis and enzymes, it governs many organic transformations like ester hydrolysis, tautomerization, and proton-coupled electron transfer.
Enzymatic covalent catalysis
Covalent catalysis in enzymes involves transient bond formation between substrate and an active-site nucleophile, creating a covalent intermediate that undergoes subsequent steps to release product. It’s essential in proteases, esterases, and many metabolic transformations for rate acceleration and specificity.
Heterogeneous catalytic hydrogenation
Heterogeneous catalytic hydrogenation occurs on metal surfaces where H2 dissociates and hydrogen atoms transfer to adsorbed substrates, saturating double or triple bonds. It’s widely used industrially for selective reductions, with activity tuned by metal, support, and reaction conditions.